









Conteúdo
Sarcoma de Ewing
O que é isso ?
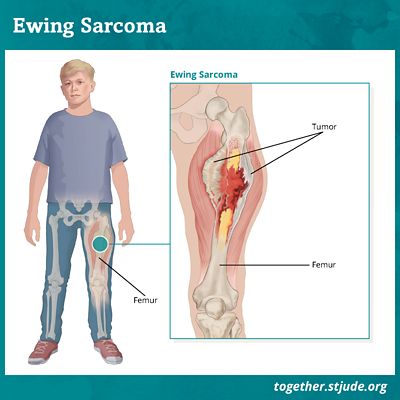
O sarcoma de Ewing é caracterizado pelo desenvolvimento de um tumor maligno nos ossos e tecidos moles. Esse tumor tem a característica de ser com alto potencial metastático. Ou a disseminação de células tumorais por todo o corpo é frequentemente identificada nesta patologia.
É uma doença rara que afeta mais geralmente as crianças. Sua incidência é de 1/312 crianças menores de 500 anos.
A faixa etária mais acometida pelo desenvolvimento dessa forma tumoral é entre 5 e 30 anos, com incidência ainda maior entre 12 e 18 anos. (3)
As manifestações clínicas associadas são dor e edema no local do tumor.
As localizações das células tumorais características do sarcoma de Ewing são múltiplas: pernas, braços, pés, mãos, tórax, pélvis, crânio, coluna, etc.
Este sarcoma de Ewing também é chamado de tumor neuroectodérmico periférico primário. (1)
Os exames médicos permitem o possível diagnóstico da doença e determinam seu estágio de evolução. O exame mais comumente associado é a biópsia.
Fatores e condições específicas podem afetar o prognóstico da doença em um indivíduo afetado. (1)
Esses fatores incluem, em particular, a disseminação de células tumorais apenas para os pulmões, cujo prognóstico é mais favorável, ou o desenvolvimento de formas metastáticas para outras partes do corpo. Neste último caso, o prognóstico é pior.
Além disso, o tamanho do tumor e a idade do indivíduo afetado têm papel fundamental no prognóstico vital. Na verdade, no caso em que o tamanho do tumor aumenta para mais de 8 cm, o prognóstico é mais preocupante. Quanto à idade, quanto mais precoce for feito o diagnóstico da patologia, melhor será o prognóstico para o paciente. (4)
O sarcoma de Ewing é um dos três principais tipos de câncer ósseo primário, juntamente com o condrossarcoma e o osteossarcoma. (2)
Sintomas
Os sintomas mais comumente associados ao sarcoma de Ewing são dor visível e inchaço nos ossos e tecidos moles afetados.
As seguintes manifestações clínicas podem ser originadas no desenvolvimento de tal sarcoma: (1)
- dor e / ou inchaço nos braços, pernas, tórax, costas ou pélvis;
- a presença de “saliências” nessas mesmas partes do corpo;
- a presença de febre sem motivo específico;
- fraturas ósseas sem motivo subjacente.
Os sintomas associados, no entanto, dependem da localização do tumor, bem como da sua importância em termos de desenvolvimento.
A dor vivenciada pelo paciente com essa patologia geralmente se intensifica com o tempo.
Outros sintomas menos comuns também podem ser visíveis, como: (2)
- febre alta e persistente;
- rigidez muscular;
- perda de peso significativa.
No entanto, um paciente com sarcoma de Ewing pode não apresentar sintomas. Nesse sentido, o tumor pode então crescer sem nenhuma manifestação clínica particular e, assim, afetar o osso ou o tecido mole sem ser visível. O risco de fratura é ainda mais importante neste último caso. (2)
As origens da doença
Como o sarcoma de Ewing é uma forma de câncer, pouco se sabe sobre a origem exata de seu desenvolvimento.
No entanto, foi apresentada uma hipótese relativa à causa do seu desenvolvimento. Na verdade, o sarcoma de Ewing afeta particularmente crianças com mais de 5 anos de idade e adolescentes. Nesse sentido, foi levantada a possibilidade de uma ligação entre o rápido crescimento ósseo nessa categoria de pessoa e o desenvolvimento do sarcoma de Ewing.
O período da puberdade em crianças e adolescentes torna os ossos e tecidos moles mais vulneráveis ao desenvolvimento de um tumor.
A pesquisa também mostrou que uma criança nascida com uma hérnia umbilical tem três vezes mais probabilidade de desenvolver o sarcoma de Ewing. (2)
Além dessas hipóteses mencionadas acima, a origem quanto à presença de uma translocação genética também foi aventada. Esta translocação envolve o gene EWSRI (22q12.2). Uma translocação t (11; 22) (q24; q12) dentro deste gene de interesse foi encontrada em quase 90% dos tumores. Além disso, muitas variantes genéticas têm sido objeto de investigações científicas, envolvendo os genes ERG, ETV1, FLI1 e NR4A3. (3)
Os fatores de risco
Do ponto de vista onde as origens exatas da patologia são, ainda hoje, pouco conhecidas, os fatores de risco também o são.
Além disso, de acordo com os resultados de estudos científicos, uma criança nascida com hérnia umbilical teria três vezes mais chances de desenvolver um tipo de câncer.
Além disso, no nível genético, a presença de translocações dentro do gene EWSRI (22q12.2) ou variantes genéticas nos genes ERG, ETV1, FLI1 e NR4A3 pode estar sujeita a fatores de risco adicionais para o desenvolvimento da doença. .
Prevenção e tratamento
O diagnóstico do sarcoma de Ewing é baseado no diagnóstico diferencial pela presença de sintomas característicos do paciente.
Após a análise do médico das áreas doloridas e inchadas, geralmente é prescrito um raio-x. Outros sistemas de imagens médicas também podem ser usados, como: Magnetic Reasoning Imaging (MRI) ou mesmo varreduras.
A biópsia óssea também pode ser feita para confirmar ou não o diagnóstico. Para isso, uma amostra de medula óssea é retirada e analisada ao microscópio. Essas técnicas diagnósticas podem ser realizadas após anestesia geral ou local.
O diagnóstico da doença deve ser realizado o mais rápido possível para que o manejo seja feito rapidamente e assim o prognóstico seja melhor.
O tratamento para o sarcoma de Ewing é semelhante ao tratamento geral para outros tipos de câncer: (2)
- a cirurgia é uma forma eficaz de tratar esse tipo de sarcoma. No entanto, a intervenção cirúrgica depende do tamanho do tumor, de sua localização e do grau de disseminação. O objetivo da cirurgia é repor a parte do osso ou tecido mole danificado pelo tumor. Para isso, uma prótese metálica ou enxerto ósseo pode ser utilizado na substituição da área afetada. Em casos extremos, a amputação de um membro às vezes é necessária para prevenir a recidiva do câncer;
- quimioterapia, geralmente usada após a cirurgia para reduzir o tumor e facilitar a cura.
- a radioterapia também é frequentemente utilizada após a quimioterapia, antes ou após a cirurgia, para reduzir o tamanho do tumor e evitar o risco de recidiva.