









Conteúdo
Vasculite de pequenos capilares
Vasculite de pequenos capilares
É um grande grupo de vasculites da parede das arteríolas, vênulas ou capilares, cujo prognóstico é muito variável dependendo se se trata de vasculite cutânea pura ou sistêmica.
O aspecto clínico mais comum é a púrpura (manchas arroxeadas que não desaparecem quando pressionadas) abaulamento e infiltrado, principalmente nos membros inferiores, agravado pela posição em pé, que pode assumir várias formas (petéquias e equimóticas, necróticas, pustulosas ...) ou livedo, formando uma espécie de malha arroxeada (livedo reticularis) ou mosqueado (livedo racemosa) nas pernas. Também podemos observar um fenômeno de Raynaud (alguns dedos ficam brancos com o frio).
Púrpura e livedo podem estar associados a outras lesões na pele (pápulas, nódulos, lesões necróticas, bolhas sangrantes), urticária fixa que não coça.
A presença de manifestações fora da pele constitui um fator de gravidade, evidenciando a presença de envolvimento vascular nos órgãos:
- dor nas articulações,
- dor abdominal, fezes pretas, distúrbio de trânsito,
- neuropatia periférica
- edema dos membros inferiores,
- Hipertensão Arterial,
- dificuldades respiratórias, asma, tosse com sangue ...
O médico prescreve exames que buscam a causa e os sinais de gravidade: exame de sangue com hemograma, pesquisa de inflamação, exames de fígado e rim, etc., pesquisa de sangue nas fezes e radiografia de acordo com os pontos de chamada ( radiografia pulmonar em caso de dificuldades respiratórias, etc.).
Vasculite desencadeada por infecção:
- bacteriana: estreptococos, cocos gram-negativos (gonococos e meningococos)
- viral: hepatite, mononucleose infecciosa, HIV, etc.
- parasita: malária ...
- fungo: Candida albicans ...
Vasculite associada a anormalidades imunológicas
- Crioglobulinemia do tipo II (monoclonal misto) e III (policlonal misto), associada a doença autoimune, infecção (especialmente hepatite C) ou doença do sangue
- Hypocomplementémie (urticarienne vascularite de Mac Duffie)
- Hyperglobulinémie (roxo hiperglobulinémique de Waldenström)
- Conectivite: lúpus, síndrome de Gougerot-Sjögren, artrite reumatóide ...
- Vasculite de doenças do sangue e malignidades
- Leucemia, linfoma, mieloma, câncer
- Vasculite associada a ANCA (anticorpos citoplasmáticos anti-neutrófilos)
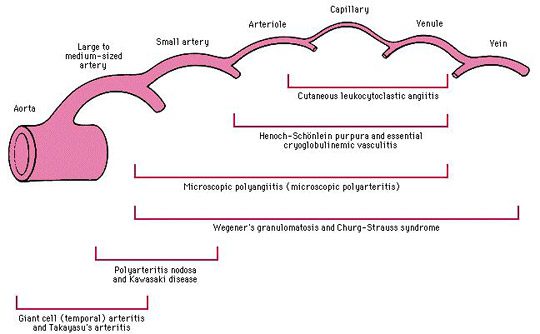
Micro Poly Angéite ou MPA
Micropoliangeíte (MPA) é uma angeíte necrosante sistêmica cujos sinais clínicos são muito semelhantes aos da PAN.
O MPA está associado ao ANCA do tipo antimieloperoxidase (anti-MPO) e geralmente dá origem a glomerulonefrite rapidamente progressiva e envolvimento pulmonar que está ausente na PAN.
O tratamento do AMF quanto à PAN começa com corticoterapia, às vezes combinada com imunossupressores (ciclofosfamida em particular)
Doença de Wegener
A granulomatose de Wegener é uma vasculite cujo início é geralmente marcado por sintomas otorrinolaringológicos ou respiratórios (sinusite, pneumopatia, etc.) resistentes a tratamentos com antibióticos.
Classicamente, o envolvimento otorrinolaringológico difuso (pansinusite destrutiva), pulmonar (nódulos parenquimatosos) e renal (glomerulonefrite necrosante pauci-imune em crescente) produz a tríade clássica da granulomatose de Wegener.
A membrana cutâneo-mucosa atinge aproximadamente 50% dos pacientes: púrpura (manchas arroxeadas que não desaparecem quando pressionadas) protuberantes e infiltradas, pápulas, nódulos subcutâneos, ulcerações cutâneas, pústulas, vesículas, gengivite hiperplásica ...
O ANCA é um teste diagnóstico e evolutivo da granulomatose de Wegener, com fluorescência citoplasmática difusa (c-ANCA), finamente granular com realce perinuclear e / ou fluorescência puramente perinuclear (p-ANCA).
O manejo da granulomatose de Wegener, que às vezes pode ser considerada uma emergência médica, deve ser realizado em ambiente hospitalar especializado, por meio de uma combinação de cortisona e ciclofosfamida oral.
Doença de Churg e Strauss
A asma é um critério principal e precoce desta vasculite, que precede em média 8 anos antes dos primeiros sinais de vasculite (neuropatia, doenças dos seios da face, etc.) e que persiste depois.
Os exames de sangue mostram, em particular, um claro aumento de leucócitos polinucleares eosinofílicos
O tratamento da doença de Churg e Strauss começa com corticoterapia, às vezes combinada com imunossupressores (especialmente ciclofosfamida)
A opinião do nosso médico
A púrpura infiltrada (manchas arroxeadas, um tanto espessas que não desaparecem com a pressão do dedo) é o principal sinal de vasculite. Infelizmente, esse sinal nem sempre está presente e a variabilidade dos sinais clínicos inespecíficos muitas vezes torna o diagnóstico difícil para os médicos. Da mesma forma, muitas vezes é difícil encontrar uma causa para tratar na vasculite de pequenos vasos, que é de longe o caso mais importante encontrado na prática atual em comparação com a vasculite de médios e grandes vasos: cerca de metade da vasculite de pequenos vasos. vasos não têm causa encontrada durante as explorações biológicas e radiológicas que o médico realiza para buscar uma etiologia. Freqüentemente falamos em “vasculite alérgica” ou “vasculite de hipersensibilidade” ou melhor, “vasculite cutânea de pequenos vasos de calibre idiopático”. Dr. Ludovic Rousseau, dermatologista |
Marcos
Grupo Francês de Estudo de Vasculite: www.vascularites.org
Dermatonet.com, site de informações sobre pele, cabelo e beleza por um dermatologista
MedicineNet: http://www.medicinenet.com/vasculitis/article.htm